


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
 molecule adsorption on polar and
molecule adsorption on polar and 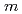 -plane surfaces of GaN
-plane surfaces of GaN角谷 正友*; 隅田 真人*; 浅井 祐哉*; 田村 亮*; 上殿 明良*; 吉越 章隆
Journal of Physical Chemistry C, 124(46), p.25282 - 25290, 2020/11
被引用回数:10 パーセンタイル:40.15(Chemistry, Physical)O分子ビームによるGaN表面[極性Ga面(+c), N面(-c)および無極性(10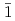 0)()面]の初期酸化をリアルタイム放射光X線光電子分光法およびDFT分子動力学計算によって調べた。三重項Oが+c面のブリッジ位置において解離または化学吸着し、N終端-c面では、O分子は解離化学吸着のみであることが分かった。面では、Oの解離吸着が支配的であるが、極性表面に吸着したO分子の結合長および角度が異なることが分かった。スピンと極性を考慮した計算モデルが金属酸化物とGaNのデバイス界面の理解に有用であることが分かった。
0)()面]の初期酸化をリアルタイム放射光X線光電子分光法およびDFT分子動力学計算によって調べた。三重項Oが+c面のブリッジ位置において解離または化学吸着し、N終端-c面では、O分子は解離化学吸着のみであることが分かった。面では、Oの解離吸着が支配的であるが、極性表面に吸着したO分子の結合長および角度が異なることが分かった。スピンと極性を考慮した計算モデルが金属酸化物とGaNのデバイス界面の理解に有用であることが分かった。
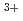 ion; A Car-Parrinello molecular dynamics study
ion; A Car-Parrinello molecular dynamics study池田 隆司; 平田 勝; 木村 貴海
Journal of Chemical Physics, 122(2), p.024510_1 - 024510_5, 2005/01
被引用回数:31 パーセンタイル:68.38(Chemistry, Physical)0.8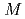 YCl
YCl 水溶液中のYイオンの水和構造と水和イオンのダイナミクスを、過剰プロトンが存在する場合としない場合の2条件について、第一原理分子動力学法により調べた。どちらの条件でもYの第一水和殻には水8分子が含まれ、それらがsquare antiprismを形成する結果となった。この構造はX線吸収端構造の実験から推測されている構造と一致している。局在軌道を用いた電子状態の詳細な解析から、水分子がYイオンに配位することにより、特に酸素原子の電荷分布に大きな変化が生じており、バルク水を基準とした
水溶液中のYイオンの水和構造と水和イオンのダイナミクスを、過剰プロトンが存在する場合としない場合の2条件について、第一原理分子動力学法により調べた。どちらの条件でもYの第一水和殻には水8分子が含まれ、それらがsquare antiprismを形成する結果となった。この構造はX線吸収端構造の実験から推測されている構造と一致している。局在軌道を用いた電子状態の詳細な解析から、水分子がYイオンに配位することにより、特に酸素原子の電荷分布に大きな変化が生じており、バルク水を基準とした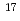 O NMRは約-20ppmの化学シフトを示すことがわかった。
O NMRは約-20ppmの化学シフトを示すことがわかった。
 molecular dynamics study of polarization effects on ionic hydration in aqueous AlCl solution
molecular dynamics study of polarization effects on ionic hydration in aqueous AlCl solution池田 隆司; 平田 勝; 木村 貴海
Journal of Chemical Physics, 119(23), p.12386 - 12392, 2003/12
被引用回数:43 パーセンタイル:78.96(Chemistry, Physical)第一原理分子動力学法を用いて、常温常圧下での0.8M AlCl水溶液中のAl及びCl の水和構造とそのダイナミクスを調べた。第一原理シミュレーションから得られたAl及びClの水和構造はどちらも実験とよく一致した。水和水の分子構造及び構成分子とイオンの双極子モーメントを詳細に検討したところ、カチオン,アニオンのどちらに対してもその水和構造とダイナミクスに分極効果が重要な役割を演じていることがわかった。Alの加水分解についても言及した。
の水和構造とそのダイナミクスを調べた。第一原理シミュレーションから得られたAl及びClの水和構造はどちらも実験とよく一致した。水和水の分子構造及び構成分子とイオンの双極子モーメントを詳細に検討したところ、カチオン,アニオンのどちらに対してもその水和構造とダイナミクスに分極効果が重要な役割を演じていることがわかった。Alの加水分解についても言及した。
町田 昌彦; 中村 博樹
no journal, ,
Thorium has been considered as an alternative nuclear fuel element to uranium. Thorium is well-known to be more abundant in nature than uranium. Furthermore, it is widely spread that thorium dioxide is chemically more stable than uranium dioxide. From an economical and secure point of view, thorium fuel can be a candidate fuel in next-generation nuclear reactors. However, the data of thermal properties of thorium dioxide are insufficient, in particular, in the high-temperature region near its melting point. In order to supplement such thermal data of the materials, classical molecular dynamics have played an important role and been applied also to thorium compounds. However, the thermal properties obtained by classical molecular dynamics often depend on the empirical parameters giving the atomic potential. Especially, it is known that the Bredig transition (rapid growth of heat capacity slightly below melting point) strongly depends on the choice of parameters of the atomic potential in the case of molecular dynamics of uranium dioxide. Thus, classical molecular dynamics is not so reliable for high-temperature behaviours, such as the Bredig transition. Therefore, we adopt first-principles molecular dynamics to avoid this parameter sensitivity. In the present paper, we evaluate thermal properties of thorium dioxide at high temperature using first-principles molecular dynamics. The calculated enthalpy agrees well with the observed data. We find that the obtained Bredig transition temperature coincides with the experimental values and that this transition is caused by the high mobility of oxygen by visualizing the oxygen motions. Consequently, we reveal that the first-principles molecular dynamics can provide reliable data of thermal properties of nuclear fuels even at high temperature.
